... Du pétrole et des idées ...
MAI 1997 N° 16
Que se passe-t-il lorsque l'on dépose une ou plusieurs molécules à la surface d'un gros agrégat d'atomes d'argon ? Les molécules migrent-elles à la surface de l'agrégat ? Se rencontrent-elles ? Le groupe de chimie théorique du SPAM a développé des outils numériques pour simuler la dynamique de ces systèmes afin de répondre à ces questions.
|
|
Les agrégats moléculaires, petits morceaux de matière condensée dont la taille peut être contrôlée, sont des objets de laboratoire très utiles en physico-chimie, par exemple pour la mise en évidence d'effets de taille dans les propriétés de la matière ou encore l'étude de la solvatation moléculaire. De façon plus inattendue, un groupe d'expérimentateurs du SPAM a montré qu'ils se comportaient comme des microréacteurs chimiques possédant des propriétés originales. L'expérience consiste à déposer séquentiellement, à la surface d'un gros agrégat d'atomes d'argon (typiquement 1000, ce qui correspond à un rayon d'environ 25 Å), un certain nombre d'espèces réactives : d'abord des molécules, telles que N2O, puis des atomes de baryum et enfin à détecter les produits de réaction par leur émission de lumière. L'une des propriétés originales des agrégats est de servir de substrat et de confiner les entités déposées à leur surface. Obligées d'évoluer dans un volume fini, elles ne peuvent s'ignorer très longtemps. La seconde est que ces agrégats, malgré leur petite taille, constituent un bain thermique. Ils sont en effet susceptibles d'absorber l'excès d'énergie fournie par la réaction chimique et permettent de mettre en évidence de nouvelles voies réactives, non observées dans les expériences de collision en phase gazeuse. Ils peuvent enfin faciliter, avant la réaction, la préparation de l'un des deux réactifs sous forme de dimère ou de trimère, et ainsi modifier profondément le mécanisme de la réaction chimique.
Les méthodes actuelles de simulation numérique permettent de répondre à bon nombre de questions sur ces systèmes et offrent en particulier la possibilité d'accéder à des données temporelles et structurales difficilement appréhendables par l'expérience. Une première étude a ainsi été réalisée sur les systèmes Ba(Ar)n et (N2O)m(Ar)n, avec m allant de 1 à 4 pour n = 125 et 147. Ces agrégats d'argon possèdent un coeur icosaédrique constitué de deux couches complètes (de 12 et 42 atomes) autour d'un atome central ainsi qu'une troisième couche, partiellement ou complètement remplie selon leur taille (c'est-à-dire, avec 70 et 92 atomes respectivement). Ces agrégats sont suffisamment volumineux pour rendre compte des effets de diffusion moléculaire et de bain thermique mais pas trop gros de manière à rester dans un domaine de temps de calcul raisonnable. Cette étude a permis de préciser la localisation des particules, leur orientation, d'apprécier leur mobilité et le rôle de la rugosité de la surface sur celle-ci et enfin leur aptitude à se regrouper pour former des dimères ou des trimères.
La simulation consiste à se donner un ensemble de particules (les atomes et les molécules) et à résoudre numériquement les équations de la dynamique classique. Au delà des problèmes techniques des simulations de dynamique moléculaire (algorithme d'intégration stable, temps de calcul raisonnable, échantillonnage pertinent), se pose le problème du potentiel d'interaction intermoléculaire lorsque l'on cherche à obtenir des données quantitatives. Le cas étudié est en effet particulièrement délicat à traiter car les interactions Ar/N2O et Ar/Ar sont du même ordre de grandeur. Le potentiel a donc été paramétré de manière à retrouver les géométries d'équilibre des complexes de van der Waals Ar-N2O, Ar-Ba et N2O-N2O connues expérimentalement. De plus, la distribution multipolaire utilisée pour calculer l'interaction électrostatique a été adaptée de façon à reproduire exactement le moment dipolaire expérimental de la molécule de N2O, afin que l'interaction dipôle-dipôle, prépondérante à longue distance, soit correctement évaluée. Cette exigence est fondamentale lorsque l'on cherche à savoir si les mouvements de deux molécules dans l'agrégat sont corrélés ou non.
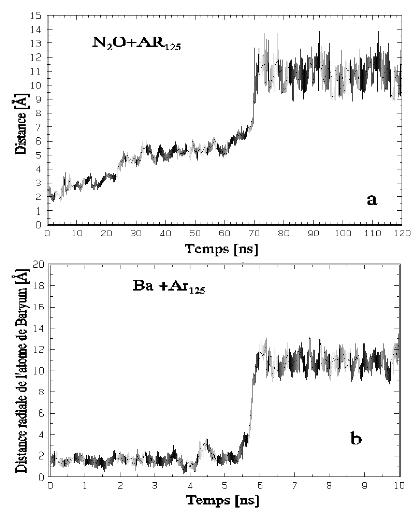
Afin de répondre à la question de la localisation, le choix a été fait de placer les molécules à l'intérieur et de regarder si elles y restaient. Dans les deux cas, Ba et N2O, la molécule sort de l'agrégat à l'échelle de temps de la dizaine de nanosecondes. La dynamique montre cependant que le processus de diffusion en volume des deux espèces est très différent. Sur la figure 1a, on observe que la molécule de N2O, qui occupe le volume de deux atomes d'argon, progresse graduellement jusqu'à la deuxième couche, puis saute enfin à la troisième. L'atome de baryum, plus volumineux, sort au contraire très brutalement de l'agrégat (Fig. 1b).
|
|
|
Les molécules de N2O en surface se déplacent beaucoup, même aux températures basses des agrégats formés en détente supersonique (typiquement 30 K). La figure 2a, représentant le déplacement par rapport à une position de référence, montre que des distances de l'ordre de la taille de l'agrégat sont déjà couvertes à l'échelle de la dizaine de nanosecondes. La molécule se trouve en surface (Fig. 2b), soit au sein de la troisième couche d'argon incomplète (R ~ 9-10 Å), soit, au dessus de celle-ci (R ~ 11 Å). Dans tous les cas, elle ne s'enfonce pas et elle s'oriente de telle sorte que ce soit l'atome d'oxygène qui émerge, ce qui ne peut que faciliter la réaction d'oxydation du baryum.
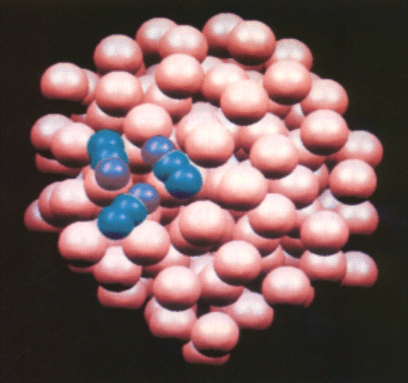
Le bain thermique évoqué plus haut est extrêmement efficace pour la formation des dimères à la surface de l'agrégat. Toutes les collisions entre deux molécules de N2O entraînent la formation d'un dimère stable et le dépôt de m (£ 4) molécules conduit à la formation de (N2O)m en surface de l'agrégat au bout de quelques dizaines de nanosecondes. La figure 3 représente une configuration instantanée d'un tel agrégat. Par ailleurs, on a montré que le coefficient de diffusion d'une molécule en surface ne dépend pas de la présence d'autres molécules : les mouvements moléculaires ne sont pas corrélés, sauf bien sûr à très courte distance. Pour trouver les temps moyens de dimérisation à la surface des gros agrégats de l'expérience (n = 400-10000), il suffit donc simplement de simuler la marche au hasard des deux particules sur une surface sphérique de rayon correspondant. On parvient ainsi à des temps typiques de l'ordre de 200 ns, ce qui permet d'adapter la chronologie de l'expérience en conséquence.
L'ensemble de ces résultats a permis de valider un modèle cohérent d'interprétation des expériences de chimie en surface d'agrégat d'argon. Mais le champ d'application de la dynamique moléculaire classique dans les agrégats est bien plus vaste. La poursuite de ce travail sera l'étude des temps caractéristiques de réorganisation d'une couche de solvant autour d'une molécule de soluté optiquement excitée, fera écho aux expériences en phase liquide réalisées sur la source laser femtoseconde du DRECAM.
Pour en savoir plus :
Sur la simulation de dynamique :
J.-P. Visticot et al., J. Chem. Phys 100,158 (1994) et
P. de Pujo et al., Z. Phys. D 25, 357 (1993)
Sur les potentiels intermoléculaires :
V. Brenner et al., Z. Phys. D 30, 327 (1994) et
Ph. Millié et al., J. Chim. Phys. 92, 428 (1995)
Sur les expériences :
A. Lallement et al., J. Chem. Phys. 99, 8705 (1993).
Contacts :
P. de Pujo, M.P. Gaigeot.
Phases Magazine N° 17
La radiolyse de l'eau ...